Воспользуйтесь поиском по сайту:
Аномалия Эбштейна
Содержание статьи:
Аномалия Эбштейна (по фамилии патологоанатома, впервые описавшего данное заболевание) – врожденный цианотический порок сердца.
При таком пороке трехстворчатый клапан располагается не в типичном месте (между предсердием и желудочком в правых отделах сердца), а намного ниже, «утоплен» вглубь полости желудочка. Объем правого желудочка существенно уменьшен за счет вдающегося клапана, предсердие же, напротив, значительно больше нормы за счет части желудочка, называющейся атриализованной, которая из-за изменения положения клапана отошла к предсердию.
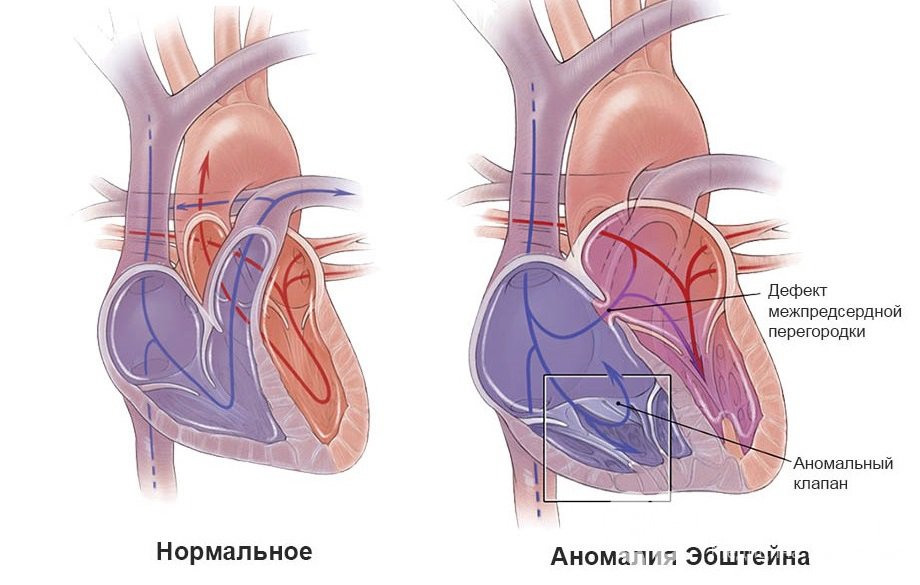 Трехстворчатый клапан при аномалии Эбштейна
Трехстворчатый клапан при аномалии ЭбштейнаПомимо изменения положения клапана, у половины пациентов с обозначенным пороком определяется дефект межпредсердной перегородки (нарушение целостности) – незаращение овального окна. Овальное окно в норме функционирует во внутриутробном периоде, в течение первых 3-5 часов жизни ребенка оно закрывается и полностью зарастает в течение 2-12 месяцев. В данном случае закрытия окна не происходит, что ведет к смешиванию венозной и артериальной крови правого и левого предсердий и, как следствие, снижению концентрации кислорода в артериальной крови. Несмотря на уменьшение эффективности кровообращения, указанный дефект зачастую оказывается жизнеспасающим, поскольку разгружает перерастянутое правое предсердие.
В отсутствие сообщения между камерами правое предсердие может достигать гигантских размеров, вмещая до 2500-3000 мл крови – при нормальном объеме до 100 мл.
Аномалия Эбштейна может протекать бессимптомно, выявляться впервые в зрелом или даже пожилом возрасте.
Также для аномалии Эбштейна характерны сращение лепестков клапана с прилежащей тканью сердца, их окончатые дефекты и провисания, а также деформация сухожильных нитей, обеспечивающих открытие и закрытие клапана.
Заболевание встречается крайне редко: на его долю приходится не более 1 случая из 100 (по некоторым данным – 200) врожденных пороков сердца.
Причины и факторы риска
Предполагается, что к возникновению аномалии Эбштейна ведут мутации в локусе хромосомы 17q CFA9, дупликация хромосомы 15q, дефект рецептора ALK3/BMPR. Хромосомные нарушения возникают на этапе слияния родительского генетического материала или на ранних сроках беременности и приводят к некорректному формированию органов и тканей организма ребенка во внутриутробном периоде.
Поскольку точные причины заболевания пока не установлены, наиболее вероятными факторами риска считаются:
- прием матерью препаратов лития во время беременности;
- прием матерью бензодиазепинов во время беременности;
- перенесенные на ранних сроках беременности вирусные заболевания (грипп, краснуха, корь);
- многократное спонтанное прерывание беременности на ранних сроках в анамнезе;
- хроническая интоксикация пестицидами, парами лакокрасочных веществ, продуктами переработки нефти и т. п. (проживание в экологически неблагополучных районах, работа на вредных производствах);
- употребление родителями запрещенных веществ, злоупотребление алкоголем, курением.
 Вирусные заболевания на ранних сроках беременности могут привести к развитию аномалии Эбштейна у плода
Вирусные заболевания на ранних сроках беременности могут привести к развитию аномалии Эбштейна у плодаФормы заболевания
Предлагается несколько классификаций типов аномалии Эбштейна, но наиболее распространена классификация E. Bacha, рассматривающая различные виды деформаций створок:
- I тип – передняя створка клапана большая и подвижная, две другие смещены, недоразвиты или отсутствуют;
- II тип – присутствуют все три створки, но они уменьшены в размере и спирально смещены по направлению к верхушке;
- III тип – ограничена подвижность передней створки, створка укорочена, сухожильные нити, приводящие ее в движение, сросшиеся и также укороченные, два других лепестка смещены и диспластичны;
- IV тип – передняя створка клапана значительно деформирована и смещена внутрь правого желудочка, его сухожильные хорды отсутствуют или присутствуют частично, задняя створка недоразвита или отсутствует, медиальная створка значительно деформирована и представлена гребневидным фиброзным выростом.
При диагностике грубых нарушений вероятность выживания новорожденного – 75% в течение первого месяца жизни. До полугода доживают 68%, до 5 лет – 64%, в последующем кривая летальности стабилизируется.
В зависимости от тяжести выделяют 3 стадии гемодинамических нарушений:
- 1-я стадия – бессимптомного течения;
- 2-я стадия – гемодинамических расстройств (2А – без нарушений сердечного ритма, 2Б – с нарушениями сердечного ритма);
- 3-я стадия – стойкой декомпенсации.
Симптомы
Клинические проявления заболевания многообразны; в основе нарушений гемодинамики лежит уменьшение объема правого желудочка. Уменьшенная в размерах камера не может вместить нормальный объем крови во время диастолы, что в итоге приводит к снижению легочного кровотока, недостаточному насыщению артериальной крови кислородом и гипоксии органов и тканей.
 Аномалия Эбштейна может выявляться в зрелом возрасте
Аномалия Эбштейна может выявляться в зрелом возрастеОсновные симптомы:
- нарушения дыхания (одышка, приступы удушья, дыхательный дискомфорт) при физическом напряжении;
- быстрая утомляемость, непереносимость нагрузок;
- приступы усиленного, «неправильного» сердцебиения;
- нарушения ритма;
- преходящая бледность или синюшное окрашивание кожных покровов и видимых слизистых оболочек;
- боли в области сердца;
- спонтанное учащение пульса;
- изменение концевых фаланг пальцев кистей (симптом барабанных палочек) и ногтей (симптом часовых стекол);
- сердечный горб (округлое, обычно симметричное выпячивание, располагающееся в передней части грудной клетки в области сердца);
- увеличение печени и селезенки.
Заболевание может протекать бессимптомно, выявляться впервые в зрелом или даже пожилом возрасте.
Особенности протекания заболевания у детей
Дети с аномалией Эбштейна рождаются с цианотичным окрашиванием кожи, которое через 2-3 месяца может уменьшиться, поскольку сопротивление сосудов легких, высокое в периоде новорожденности, снижается. Но если дефект в перегородке небольшой или отсутствует, состояние половины детей в этот период становится критическим, и они могут погибнуть от нарастающей сердечной недостаточности и осложнений цианоза уже в первые недели жизни.
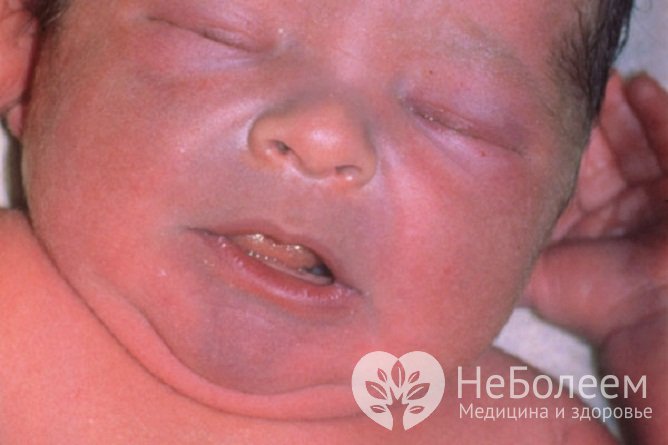 Дети с аномалией Эбштейна рождаются с цианотичным окрашиванием кожи
Дети с аномалией Эбштейна рождаются с цианотичным окрашиванием кожиЧитайте также:5 загадочных недугов, причины которых неизвестны медицине
Диагностика
При объективном обследовании сердечно-сосудистой системы определяются:
- расширение границ сердечной тупости вправо и влево;
- глухие, ослабленные тоны сердца, часто выслушивается ритм галопа, то есть трех- или четырехчленный ритм, обусловленный раздвоением I и II тонов сердца или наличием дополнительных III и IV тонов.
Аномалия Эбштейна встречается крайне редко: на его долю приходится не более 1 случая из 100 (по некоторым данным – 200) врожденных пороков сердца.
Данные инструментальных методов исследования следующие:
- при ЭКГ низкого вольтажа – выраженные остроконечные зубцы Р, которые указывают на гипертрофию и дилатацию правого предсердия, блокада правой ножки пучка Гиса, признаки синдрома Вольфа – Паркинсона – Уайта (WPW);
- при рентгенографии – кардиомегалия, снижение интенсивности легочного рисунка, в боковой проекции – аномальное наполнение ретростернального пространства;
- при УЗ-исследовании сердца – удлинение, утолщение и провисание лепестков трехстворчатого клапана, расширение правого предсердия, при допплерографии – характерный «воющий» звук движения створок;
- при катетеризации сердца (производится в редких случаях) – повышенное давление в правом предсердии;
- при ангиокардиографии – гигантская, резко расширенная полость правого предсердия с высокой интенсивностью контрастирования.
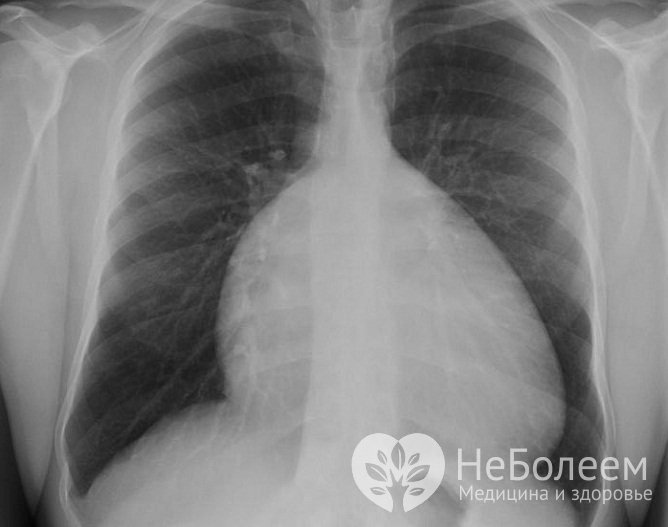 Рентгеновский снимок аномалии Эбштейна
Рентгеновский снимок аномалии ЭбштейнаЛечение
Основным способом радикального устранения аномалии Эбштейна является хирургическое вмешательство, которое может осуществляться в один или несколько этапов.
У 90% пациентов с аномалией Эбштейна после хирургического лечения отмечаются положительные непосредственные и отдаленные результаты.
Показания к хирургическому лечению:
- сердечная недостаточность III–IV ФК (функциональных классов);
- значительный или прогрессирующий цианоз – уровень насыщения крови кислородом (индекс сатурации) менее 80%;
- тяжелая кардиомегалия с кардиоторакальным индексом больше 0,65;
- сопутствующие аномалии сердца;
- предсердные и желудочковые аритмии;
- наличие парадоксальной эмболии в анамнезе.
Реконструктивные операции включают исправление трикуспидального клапана, его перемещение, замещение (протезирование), закрытие предсердной перегородки, пластику атриализированной части правого желудочка.
 Хирургическое вмешательства – основной метод лечение аномалии Эбштейна
Хирургическое вмешательства – основной метод лечение аномалии ЭбштейнаХирургическое лечение улучает выживаемость, прогноз, предупреждает развитие или существенно уменьшает выраженность аритмий.
Возможные осложнения и последствия
Наиболее распространенные последствия аномалии:
- инфекционный эндокардит;
- сердечная недостаточность;
- острые жизнеугрожающие нарушения ритма сердца;
- внезапная сердечная смерть;
- абсцесс мозга;
- острое нарушение мозгового кровообращения;
- транзиторные ишемические атаки;
- парадоксальная эмболия.
Прогноз
Ранний дебют заболевания в детском или юношеском возрасте – прогностически неблагоприятный признак.
При диагностике грубых нарушений вероятность выживания новорожденного – 75% в течение первого месяца жизни. До полугода доживают 68%, до 5 лет – 64%, в последующем кривая летальности стабилизируется.
Вероятность смертельного исхода при оперативном вмешательстве зависит от тяжести аномалии в каждом конкретном случае, наличия сопутствующей патологии. У 90% пациентов после хирургического лечения отмечаются положительные непосредственные и отдаленные результаты. В течение года возможно восстановление трудоспособности.
Об авторе

Информация является обобщенной и предоставляется в ознакомительных целях. При первых признаках болезни обратитесь к врачу. Самолечение опасно для здоровья!
Существуют очень любопытные медицинские синдромы, например, навязчивое заглатывание предметов. В желудке одной пациентки, страдающей от этой мании, было обнаружено 2500 инородных предметов.






















Нашли ошибку в тексте? Выделите ее и нажмите Ctrl + Enter ООО «Медицинская практика», ОГРН 1187456030354